Research Article - Der Pharmacia Lettre ( 2023) Volume 15, Issue 7
Received: 29-Jun-2023, Manuscript No. DPL-23-56851;
Editor assigned: 03-Jul-2023, Pre QC No. DPL-23-56851 (PQ);
Reviewed: 17-Jul-2023, QC No. DPL-23-56851;
Revised: 24-Jul-2023, Manuscript No. DPL-23-56851 (R);
Published:
31-Jul-2023
, DOI: 10.37532/ 0975-5071.2023.15.06
, Citations: Vinayak V. 2023. Environmentally Benign Synthesis of 5-Arylidene-Rhodanine Derivatives in Room Temperature
Diisopropyl Ethyl Ammonium Acetate. Der Pharma Lett.15:06-15
,
Copyright: © 2023 Vinayak V. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited
The Di-Isopropyl Ethyl Ammonium Acetate (DIPEAc) mediated Knoevenagel condensation protocol has been established for the first time by a successive reaction of aldehydes and rhodanine to afford biologically active 5-arylidene-rhodanine derivatives in high to excellent yields at room temperature. The key benefits of the present protocol are the capability to allow a diverse functional groups, high yields, easy workup, short reaction times, recyclability of the catalyst and solvent free conditions, thus providing environmentally benign protocol.
Room temperature ionic liquids, DIPEAc, Environmentally benign, 5-Arylidene-rhodanine, Knoevenagel condensation.
Room Temperature Ionic Liquids (RTILs) have taken the attention of the research community all over the globe as a green alternative route to traditional ecofriendly media for synthesis, catalysis, separation and other several chemical tasks [1-4]. RTILs include diverse properties, such as non-volatility, extensive liquid range, high thermal stability, low toxicity, excellent solubility, noncombustible and recyclability [5]. RTILs act as “neoteric solvents” for a broad range of chemical and industrial processes. In recent times, RTILs have been originating to be valuable as environmental friendly media for countless organic revolutions [6,7]. Thus, the introduction of a dynamic, mild, inexpensive and environmental benign catalyst for significant cyclization reaction superior to analogues of biological and pharmaceutical distinction is in demand. In this paper, we have validated DIPEAc as ionic liquid catalyzed the proficient synthesis of 5-arylidene-rhodanine derivatives under ecofriendly reaction conditions (Figures 1 and 2).

Figure 1. Synthesis of 5-Arylidene-rhodanine derivatives 3a by using DIPEAc as RTIL.

Figure 2. Synthesis of Di-Isopropyl Ethyl Ammonium Acetate (DIPEAc).
Rhodanine (2-thioxothiazolid-4-one) and their analogues have been known as advantaged structures in pharmacological research and exhibits broad spectrum of biological activities [8]. Particularly, 5-arylidene rhodanines display antibacterial, antifungal, antinflammatory, antitubercular activity and DNA polymerase λ inhibitors. In addition to this, chikungunya virus, murd ligase, dengue virus protease, Alzheimer’s disease, tau aggregation, HIV inhibitors, GTPase activity, epalrestat as a aldose reductase inhibitors, histone acetyltransferases, AF6 PDZ domain, HCV NS5b, Bcl-2, PDE4, JNK Stimulating Phosphatase-1 (JSP-1), aldose reductase, ADAMTS-5, trypanosoma brucei Dolichol Phosphate Mannose Synthase (DPMS), photosynthesis, anthrax lethal factor protease, fungal protein mannosyl transferase-1, protease, β-lactamase and HCV NS3 protease inhibitor (Figure 3) [9,10].

Figure 3. Arylidene-rhodanine incorporated bioactive molecules.
Owing to the latent significance of rhodanine as a key moiety in life sciences, pharmaceuticals, wide reaching hard work have made in the last few decades by researchers and numerous protocols have been developed for their synthesis using various catalysts such as pyridine ethanol, piperidine ethanol/acetic acid, NH4OAc/AcOH, CH3COONa/AcOH, NH4OAc/toluene, NH4Cl/NH4OH-EtOH, NH4OAc and (bmim) (OH). However, several of these testify methods become contaminated with numerous disadvantages such as use of hazardous or costly reagents, strong acidic conditions, and low yields of products, long reaction times and sophisticated treatment. Moreover, many of these figures utilize organic solvents as the reaction medium. Hence, competent, innovative and ecofriendly procedures are still powerfully needed to generate biologically active molecules. As per our examination, the existential of this work is to start on a speedy and well organized synthetic procedure for obtaining xanthene derivatives under ecological conditions. As an extension of emerging efficient and economic strategy to develop biologically and pharmaceutically important molecules, herein, we reported a first time two component method in DIPEAc at room temperature to access a library of arylidene-rhodanine derivatives in good to excellent yields [11,12].
All of the reagents used were of laboratory grade. Melting points of all of the synthesized analogues were resolute in an open capillary tube and are uncorrected. The progress of the reaction was monitored by thin layer chromatography on Merck’s silica plates, and imagining was accomplished by iodine/ultraviolet light. 1H NMR spectra were recorded with a Bruker AvIII HD-400 MHz spectrometer operating at 400 MHz using DMSO solvent and Tetramethylsilane (TMS) as the internal standard and chemical shift in δ ppm. Mass spectra were recorded on a waters UPLCTQD (ESI-MS and APCI-MS) instrument and elemental analysis was recorded on the CHNS auto analyzer (Thermo Fischer EA1112 series). Chemical shifts (δ) are reported in parts per million using TMS as an internal standard. The splitting pattern abbreviations are designed as singlet (s); doublet (d); double doublet (dd); bs (broad singlet), triplet (t); quartet (q); and multiplets (m) [13,14].
General procedure for the synthesis of Di-Isopropyl Ethyl Ammonium Acetate (DIPEAc). A mixture of N,N-diisopropylethylamine (3 mmol) and acetic acid (3 mmol) was stirred at 0-10°C for 20 min. The viscous liquid, diisopropylethylammonium acetate, was achieved.
General procedure for synthesis of (Z)-5-benzylidene-2-thioxothiazolidin-4-one (3a-h)
A mixture of aryl aldehyde (1 mmol), rhodanine (1 mmol) and ionic liquid DIPEAc (4 mL) was stirred at room temperature for 40 min. The progress of the reaction was monitored by thin layer chromatography (n-Hexane/EtOAc 8:2). Further, addition of ice cold water (10 mL) was added to it and stirred for 10 min and filtered. The obtained solid was filtered, washed with cold water to remove the ionic liquid. The obtained crude compounds were recrystallized using ethanol ethyl acetate [15,16].
(Z)-5-benzylidene-2-thioxothiazolidin-4-one (3a)
The compound 3a was obtained via Knoevenagel condensation reaction between 1a and 2a as yellow solid: Mp: 196-198οC; yield: 93%; 1H NMR (400 MHz, DMSO-d6): δ 9.38 (s, NH, 1H), 7.74 (s, Ar-CH=C, 1H), 7.27 (t, J=7.6 Hz, 2H), 7.21-7.09 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 195.17, 160.82, 144.43, 135.24, 128.02, 114.49 and 112.18.
(Z)-5-(3-methylbenzylidene)-2-thioxothiazolidin-4-one (3b)
The compound 3b was obtained via Knoevenagel condensation reaction between 1b and 2a as yellow solid; Mp: 208-210οC; Yield: 87%; 1H NMR (400 MHz, DMSO-d6): δ 9.20 (s, NH, 1H), 7.75 (s, Ar-CH=C, 1H), 7.13-6.95 (m, 3H), 6.86 (d, J=6.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 197.41, 168.12, 141.32, 133.21, 129.97, 128.66, 126.27, 123.66, 122.27 and 23.31.
(Z)-5-(4-methylbenzylidene)-2-thioxothiazolidin-4-one (3c)
The compound 3c was obtained via Knoevenagel condensation reaction between 1c and 2a as yellow solid; Mp: 230-232οC; Yield: 92%; 1H NMR (400 MHz, DMSO-d6): δ 9.54 (s, NH, 1H), 7.78 (s, Ar-CH=C, 1H), 7.14 (d, J=7.8 Hz, 2H), 6.98 (d, J=7.5 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 196.36, 162.17, 141.21, 135.61, 128.71, 128.20, 115.67 and 21.02.
(Z)-5-(3-methoxybenzylidene)-2-thioxothiazolidin-4-one (3d)
The compound 3d was obtained via Knoevenagel condensation reaction between 1d and 2a as yellow solid; Mp: 162-164οC; Yield: 89%; 1H NMR (400 MHz, DMSO-d6): δ 9.56 (s, NH, 1H), 7.65 (s, Ar-CH=C, 1H), 7.03 (t, J=8.1 Hz,1H), 6.79 (d, J=6.9 Hz, 2H), 6.56 (d, J=7.0 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 196.26, 162.35, 159.26, 145.68, 128.77, 120.72, 115.42, 114.26, 111.78 and 53.2.
(Z)-5-(4-methoxybenzylidene)-2-thioxothiazolidin-4-one (3e)
The compound 3e was obtained via Knoevenagel condensation reaction between 1e and 2a as yellow solid; Mp: 260-262οC; Yield: 91%; 1H NMR (400 MHz, DMSO-d6): δ 9.60 (s, NH, 1H), 7.76 (s, Ar-CH=C, 1H), 7.23-7.09 (m, 2H), 6.70 (dd, J=5.8, 2.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 196.40, 162.05, 157.90, 136.47, 129.25, 115.72 and 54.5.
(Z)-5-(3,4-dimethoxybenzylidene)-2-thioxothiazolidin-4-one (3f)
The compound 3f was obtained via Knoevenagel condensation reaction between 1f and 2a as yellow solid; Mp: 200-202οC; Yield: 88%; 1H NMR (400 MHz, DMSO-d6): δ 9.65 (s, NH, 1H), 7.78 (s, Ar-CH=C,1H), 6.88 (s, 1H), 6.71 (td, J=8.2, 4.3 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ190.85, 168.53, 154.64, 152.87, 141.68, 132.62, 127.24, 126.61, 55.48 and 54.61.
(Z)-5-(3-nitrobenzylidene)-2-thioxothiazolidin-4-one (3g)
The compound 3g was obtained via Knoevenagel condensation reaction between 1g and 2a as red solid; Mp: 172-174οC; yield: 84%; 1H NMR (400 MHz, DMSO-d6): δ 9.57 (s, NH, 1H), 8.06–7.90 (m, 2H), 7.78 (d, J=6.7 Hz, 1H), 7.65 (s, Ar-CH=C, 1H), 7.39 (d, J=7.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 197.42, 167.96, 147.56, 141.40, 131.53, 128.54, 127.28 and 125.82.
(Z)-5-(3-iodobenzylidene)-2-thioxothiazolidin-4-one (3h)
The compound 3h was obtained via Knoevenagel condensation reaction between 1h and 2a as pale yellow solid; Mp: 276-278οC; yield: 85%; 1H NMR (400 MHz, DMSO-d6): δ 9.70 (s, NH, 1H), 7.78 (s, Ar-CH=C, 1H), 7.53 (s, 1H), 7.41 (d, J=7.8 Hz, 1H), 7.31 (d, J=7.6 Hz, 1H), 6.94 (t, J=7.8 Hz, 1H); 13C NMR (100 MHz, DMSO-d6): δ 191.01, 165.83, 140.46, 132.57, 129.02, 127.61, 127.36 and 123.87.
(Z)-5-(4-bromobenzylidene)-2-thioxothiazolidin-4-one (3i)
The compound 3i was obtained via Knoevenagel condensation reaction between 1i and 2a as red solid; Mp: 232-234οC; yield: 88%; 1H NMR (400 MHz, DMSO-d6): δ 9.42 (s, NH, 1H), 7.75 (s, Ar-CH=C, 1H), 7.33 (d, J=6.9 Hz, 2H), 7.17 (d, J=6.8 Hz, 2H), 4.69 (s, 1H), 2.46 (s, 4H), 2.20 (q, J=16.4 Hz, 4H), 1.10 (s, 6H), 0.98 (s, 6H); 13C NMR (100 MHz, DMSO-d6): δ 196.26, 162.41, 143.18, 131.09, 130.13, 120.18 and 115.14.
(Z)-5-(4-chlorobenzylidene)-2-thioxothiazolidin-4-one (3j)
The compound 3j was obtained via Knoevenagel condensation reaction between 1j and 2a as yellow solid; Mp: 228-230οC; Yield: 92%; 1H NMR (400 MHz, DMSO-d6): δ 9.63 (s, NH, 1H), 7.78 (s, Ar-CH=C, 1H), 7.24 (d, J=8.4 Hz, 2H), 7.21–7.14 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 196.28, 162.44, 142.70, 131.93, 129.74, 128.13 and 115.18.
(Z)-5-(4-fluorobenzylidene)-2-thioxothiazolidin-4-one (3k)
The compound 3k was obtained via Knoevenagel condensation reaction between 1k and 2a as red solid; Mp: 226-228οC; Yield: 91%; 1H NMR (400 MHz, DMSO-d6): δ 9.78 (s, NH, 1H), 7.78 (s, Ar-CH=C, 1H), 7.31 (s, 1H), 7.06 (d, J=7.8 Hz, 2H), 6.54 (d, J=7.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ 197.30, 162.45, 154.70, 135.47, 129.29, 115.85 and 115.23.
(Z)-5-(4-nitrobenzylidene)-2-thioxothiazolidin-4-one (3l)
The compound 3l was obtained via Knoevenagel condensation reaction between 1l and 2a as white solid; Mp: 254-256οC; Yield: 81%; 1H NMR (400 MHz, DMSO-d6): 7.66 (s, 1H, =CH, 1H), 7.36 (d, 2H, Ar-H), 6.91 (d, 2H, Ar-H) and 4.79 (s, 2H, CH2); 13CNMR (100 MHz, DMSO-d6): δ and 190.7, 163.9, 163.3, 136.4, 132.1, 129.3, 129.0, 128.5 and 125.6.

Figure 4. 1H and 13C NMR of representative compounds.
Chemistry
To attain optimized conditions procedure based on the reaction of benzaldehyde (1a) (1 mmol), and rhodanine (2) (1 mmol) as model substrates, we checked changed temperatures, solvents and catalysts and the results of this study are summarize in Table 1. It was found that when the reaction was carried out in the absence of the catalyst in ethanol, no product was apparent, even after 8 h (Table 1, entry 1). To obtain the chosen product (3a), we tested the reaction using diverse catalysts such as Cs2CO3, p-TSA, β-CD, CTAB, SDS, ChCl: 2urea, ChCl: 2ZnCl2, PEG-400 and DIPEAc (Table 1, entries 2-11). Thus, room temperature DIPEAc as the pre-eminent catalyst was tested for this reaction. In the presence of DIPEAc, compound 3a was isolated in 93% yield after only 40 min at room temperature. The model reaction in water using phase transfer catalysts is found to be sluggish and formed the desired 3a in less yields. Therefore, it can be thought that DIPEAc is green and a better solvent and catalyst compared to the others shown in Table 1 [17-19].
| Entry | Catalyst | Medium | Time | Yieldb (%)/Time (h) |
|---|---|---|---|---|
| 1 | - | EtOH | 8 h | Trace |
| 2 | Cs2CO3 | EtOH | 6 h | 61 |
| 3 | p-TSA | H2O | 6 h | 65 |
| 4 | β-CD | H2O | 6 h | 62 |
| 5 | CTAB | H2O | 5 h | 58 |
| 6 | SDS | H2O | 5 h | 52 |
| 7 | ChCl:2ZnCl2 | ChCl: 2ZnCl2 | 3 h | 75 |
| 8 | ChCl:2urea | ChCl: 2urea | 2 h | 80 |
| 9 | PEG-400 | PEG-400 | 5 h | 72 |
| 10 | DIPEAc | DIPEAc | 40 min | 93 |
a=Reaction conditions: Aldehyde (1 mmol), rhodanine (1 mmol) in medium (4 mL), stirred at room temperature. b=Isolated yields.
Table 1: Efficiency comparison of various catalysts for the synthesis of arylidene-rhodanine derivatives.
The amount of the catalyst is another critical parameter in terms of reaction efficiency. To confirm the amount of the DIPEAc, the model reaction was examined by a set of experiments by the varying amounts from 1 to 5 mL; as the amount of DIPEAc increases gradually, a steady increase was observed in the product yield. DIPEAc (4 mL) furnishes 3a in 93% yield at room temperature (Table 2, entries 4). Further increase in the amount of DIPEAc does not increase in the yield of the product. The model reaction was carried out without any catalyst and solvent; the trace amount of the product was achieved after a long period (Table 2, entry 1). Further, the efficiency of DIPEAc was checked by using 20 mol% DIPEAc in various solvents (Table 2, entries 7-12). In ethanol, the reaction takes place smoothly with high yield. While in water, MeOH, acetonitrile, DCM, CH2Cl2, and DMF reaction proceeds with lower yields at reflux temperature. None of the solvents exist the advantage of time and yield over the solvent free condition. Hence, the solvent free condition was regarded as the finest for the cost and environmental suitability.
| Entry | DIPEAc | Temp (°C) | Solvent | Time (min) | Yield (%)b |
|---|---|---|---|---|---|
| 1 | 0 | RT | - | 24 h | trace |
| 2 | 1 mL | RT | - | 40 | 58 |
| 3 | 2 mL | RT | - | 40 | 76 |
| 4 | 3 mL | RT | - | 40 | 82 |
| 5 | 4 mL | RT | - | 40 | 93 |
| 6 | 5 mL | RT | - | 40 | 91 |
| 7 | 20 mol% | reflux | H2O | 6 h | 72 |
| 8 | 20 mol% | reflux | EtOH | 6 h | 80 |
| 9 | 20 mol% | reflux | MeOH | 6 h | 74 |
| 10 | 20 mol% | reflux | CH3CN | 6 h | 56 |
| 11 | 20 mol% | reflux | CH2Cl2 | 6 h | 50 |
| 12 | 20 mol% | reflux | DMF | 6 h | 52 |
a=Reaction conditions: Aldehyde (1 mmol), rhodanine (1 mmol) in solvent (4 mL), stirred at room temp. b=Isolated yields.
Table 2: Solvent effects on the reaction of aldehyde and rhodanine for the synthesis of xanthene derivatives (3a).
extremely superlative method to economic and greener preparation is recovery and recyclability of an ionic liquid. Therefore we have to check the efficiency of catalyst after recover from the reaction media during the work up procedure. When reaction is completed, then reaction mass was pour on ice cold water to obtained fine crystal of final arylidene-rhodanine derivatives. In the last step removal H2O from filtrate using reduced pressure to give viscous liquid, which is on cooling to give pure ionic liquid. Recovered catalysts were reused for next four repeated cycles without considerable loss in catalytic efficiency (Table 3).
| Entry | Run | Time a (min) | Yield b |
|---|---|---|---|
| 1 | fresh | 40 | 93 |
| 2 | 2 | 40 | 91 |
| 3 | 3 | 40 | 87 |
| 4 | 4 | 40 | 85 |
| 5 | 5 | 40 | 82 |
a=Reaction progress monitored by TLC. b=Isolated yield.
Table 3: Reusability of DIPEAc ionic liquid for model reaction.
In summary, the maximum competence and greatest response time for the model reaction was observed at room temperature by using 4 mL of DIPEAc. Having ideal conditions in hand, the adaptableness of the procedure was examined for the creation of rhodanine derivatives (3a-l). The scope and efficiency of this method was explored under the optimized conditions. For this, a broad range of structurally diverse aromatic aldehydes were successfully condensed with rhodanine 2a using DIPEAc as a catalyst under solvent free conditions at room temperature ºC (Figure 5) and the obtained results are displayed in Table 4. The results clearly reveals that the condensation reactions using DIPEAc catalyst shows an extraordinary and exceptional presentation irrespective to the attendance of an electron donating/withdrawing groups on the aromatic aldehydes (Table 4) and hence this protocol is extremely effectual, talented and universal for the synthesis of 5-arylidene-rhodanine conjugates. All the synthesized compounds 3b-l was well characterized by advanced spectroscopic techniques 1H NMR and 13C NMR analysis (Figure 5).

Figure 5. synthesis of 5-arylidene-rhodanine conjugates.
• Easily available starting materials.
• DIPEAc as a greener and environmentally friendly catalyst.
• Reusable and recyclable catalyst without loss in catalytic activity.
• Synthesis of 5-arylidene-rhodanine derivatives.
• Room temperature ionic liquids.
| Compound | Structure | Yieldsb (%) | Mp ºC (observed)c | Mp ºC (reported) |
|---|---|---|---|---|
| 3a | 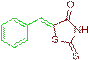 |
93 | 196-198 | 204-206 |
| 3b | 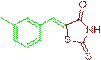 |
87 | 208-210 | - |
| 3c | 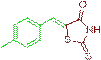 |
92 | 230-232 | 232-234 |
| 3d | 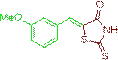 |
89 | 162-164 | - |
| 3e | 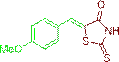 |
91 | 260-262 | 260-261 |
| 3f | 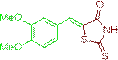 |
88 | 272-274 | 271-273 |
| 3g | 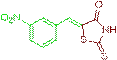 |
84 | 172-174 | - |
| 3h | 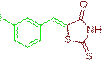 |
85 | 276-278 | - |
| 3i | 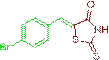 |
88 | 232-234 | 230-232 |
| 3j | 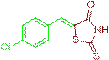 |
92 | 228-230 | 232-233 |
| 3k | 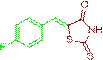 |
91 | 226-228 | 226-228 |
| 3l | 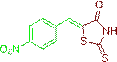 |
81 | 254-256 | 255-257 |
| a=Reaction conditions: Aldehydes (1a-l) (1 mmol) and rhodanine (2) (1 mmol) in DIPEAc (4 mL) stirred at room temp; b=isolated yields, c=melting points are in good contact with those reported in the literature. | ||||
Table 4: Synthesis of 5-arylidene-rhodanine derivatives
The structure of the titled product 3a was confirmed by 1H NMR and 13C NMR. In 1H NMR spectra of compound 3a exhibit singlet bands for methylidine proton at δ 7.74 ppm. The -NH protons of the rhodanine ring observed at δ 9.38 ppm suggest that rhodanine ring in the final compound. In the 13C NMR spectrum of compound 3a, two distinct characteristic signals exhibited at δ 160.8 and 195.2 ppm due to the -N-C=O and -S-C=S groups, respectively. Hence according these two signals suggest that formation of rhodanine ring in the final compound 3a.
A plausible mechanism for the synthesis of 5-arylidene-rhodanine derivatives via Knoevenagel condensation catalyzed by DIPEAc is shown in Figure 6. As a DIPEAc is a protic ionic liquid, initially aldehyde 1a was protonated to form intermediate II followed by nucleophilic attack of the active methylene compound 2a produces intermediate III via C-C bond formation. The final compound 3a was formed by the E1CB elimination (removal of H2O) from intermediate IV and regeneration of ionic liquid i takes place.

Figure 6. Plausible reaction mechanism pathway for the synthesis of (Z)-5-benzylidene-2-thioxothiazolidin-4-one (3a).
In conclusion, an environmentally and highly efficient green methodology has been established for the synthesis of functionalized 5-arylidene-rhodanine derivatives using an inexpensive and recoverable room temperature DIPEAc catalytic solvent free system. This, to the best of our knowledge, has no examples. This reaction Figure exposes a number of advantages, such as uniqueness, high atom efficiency, mild reaction conditions, clean reaction profiles, easy workup procedure and eco-friend liness. Furthermore, the prevention of hazardous organic solvents during the entire procedure (synthesis, ionic liquid preparation, and work up procedure) makes it a convenient and attractive method for the synthesis of these important compounds.
There are no conflicts to declare.
The authors VV are thankful to the head, department of chemistry, Dr. Shri Shivaji college Omerga, India for providing laboratory facility.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
Citation: Vinayak V. 2023. Environmentally Benign Synthesis of 5-Arylidene-Rhodanine Derivatives in Room Temperature Diisopropyl Ethyl Ammonium Acetate. Der Pharma Lett.15:06-15
Copyright: © 2023 Vinayak V. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.