Research Article - Journal of Computational Methods in Molecular Design ( 2018) Volume 8, Issue 1
“In silico” molecular docking is a new theoretical approach in pharmaceutical chemistry that allows the prediction of the most favorable mode of interaction of a ligand within its receptor (therapeutic target) in a limited time and especially sometimes without having to synthesize these compounds. In our research, the molecular docking program, Surflex, has been used to develop in silico new inhibitors of class C Beta-Lactamase deactivating several beta-lactam antibiotics including Cephalosporins. By tri-substitution of a compound “GLB”, we were able to improve significantly its score from 4.51 to 7.91 by introducing a hydroxyl group at the C8b position, a carboxyl group at the C12 position and a benzene-3’-COOH ring at its C3 carbon. The application of the Lipinski rule allowed us to verify the bioavailability of the compound resulting from the tri-substitution that is considered as a new powerful theoretical inhibitor and selective for class C Beta-lactamase.
Molecular docking, Beta-Lactamase, Therapeutic target, Surflex, score, Tri-substitution, Lipinski rule
BLSE: Extended spectrum Beta-Lactamase; AmpC: Cephalosporinase coded by ampC gene; PDB: Protein Data Bank; IC: Inhibitory Concentration; ADME: Absorption, Distribution, Metabolism, Elimination; GLB: Glycylboronate; VMD: Visual Molecular Dynamics
The excessive and non-regulated use of antibiotics has led to an extended propagation of the resistance to these molecules and the appearance of resisting microbial strains [1] especially in the countries where antibiotics are delivered without prescriptions or countries lacking standardized therapeutic guidelines where they are excessively prescribed. In such conditions, bacteria have been developed mechanisms to avoid the fatal action of antibiotics. Thus, we are in presence of resistance [2]. In fact, the production of Beta-Lactamase is the resistance mechanism the most frequent and nowadays more than 850 Beta-Lactamases are identified. These enzymes catalyze the cleavage of the amide bond of the Beta-Lactam ring through a Serine residue of their active site, which causes the irreversible opening of this ring. The products obtained are completely devoid of their antibacterial activity. Therefore, Beta- Lactamases threaten the activity of beta-lactam antibiotics [3].
To oppose this affecting enzymatic process, it is almost imperative to associate a compound to the antibacterial agent in order to inhibit the concerned enzyme [4]. The combination Beta-Lactam-Inhibitor of Beta-Lactamase allowed the evolution of the ability to cure several types of common infections, nosocomial infections and polymicrobial infections [5]. However, the developed Beta-Lactamase inhibitors recognize, nowadays, resistance cases and their number is continuously increasing [6]. The class C Beta-Lactamases is Cephaloporinases clinically important and coded by a chromosome, which has lot of Enterobacteria. They confer a resistance to Cephalosporin, penicillin and to a lot of Beta-Lactamase inhibitors [7,8].
In order to develop new potent class C Beta-Lactamase inhibitors, we used the molecular docking method. It is a computational method which aims to simulate and predict the binding affinities and the interaction modes of ligands for a therapeutic target.
In our work, we used the molecular docking’s program Surflex V 1.3 to analyze the binding mode of the compound Glycylboronate (GLB-compound) which is considered as one of the best Beta-Lactamase inhibitors. In the perspective of increasing its binding affinity, the structure of GLB-compound was taken and several types of substitutions were carried on several positions. The resulting new Beta-lactamase inhibitors may constitute good leads for Betalactamase’s drug design.
Preparation of the ligands
The Beta-Lactamase inhibitors have been drawn using Titan program taking into account the hybridization status of each atom. Subsequently, a step of energy’s minimizing is necessary in order to optimize geometrically the drawn molecules. In this context, we used the semi empirical method Austin Model 1 with the MMFF force field. The total charge of the compounds was kept as neutral. The others parameters were used in their default mode. Finally, the obtained ligands are exported in format. pdb.
Preparation of the receptor
The 3D structure of the Beta-Lactamase; subject of our study, has been acquired from the database Protein Data Bank under the code 3S1Y. This choice is dictated by its weak resolution value of 1.74 Ǻ. This code corresponds to the 3D structure of the Beta-Lactamase in complex with a powerful inhibitor that has the code S1Ywith IC50=600 nM [9].
As 3S1Y is a homodimeric enzyme, we have eliminated the chain B of the Beta-Lactamase and have kept just the chain A for an easy use of the enzyme for molecular docking. Likewise, the water molecules and the different ligands have been eliminated. Hydrogens and missing heavy atoms were added to the receptor structure followed by minimization step using YASARA force field [10]. Thus, the prepared enzyme is exported in a .mol2 file format.
The protomol generation
The molecular docking program Surflex v 1.3 uses the Hammerhead algorithm modified to realize the semi-flexible docking of the ligands in the active site of a given target. First, it generates an ideal pseudo-molecule to interact with the studied bond site, commonly known as «protomol». It consists of a set of hydrophobic and hydrophilic probes (CH4, NH and CO) that completely fit the cavity surface, making all possible interactions with the binding site residues [11,12]. The generation of the protomol was undertaking using the default parameters of Surflex according to the following command:
surflex-dock proto ligand.pdb proteine.mol2 pl
The molecular docking
The molecular docking through Surflex program allows having the best ten (10) positions (conformations) of each ligand in the studied active site, classified by their score. This step has been realized by the following command:
surflex-dock dock ligand.pdb pl-protomol.mol2 proteine.mol2
Lipinski rule
« The rule of 5 » commonly known as: «Lipinski rule » is a rule of thumb to determine if a chemical compound with a certain pharmacological or biological activity has properties that would make it a likely orally active drug in humans. [13].
According to this rule, a compound can be given orally only if it fulfills 3 out of the following 5 criteria:
• The molecular weight of the compound should not be greater than 500 Da;
• The decimal logarithm of the Octanol/water (log/P) partition coefficient should be less or equal to five (≤ 5);
• The number of the hydrogen bonding donors (grouping OH and NH : n OHN) should be less or equal to five (≤ 5);
• The number of the hydrogen bonding acceptors (atoms O and N : n ON) should be less or equal to ten ( ≤ 10);
• The number of the rotable bonds (flexible and linear) should be less or equal to fifteen (≤ 15).
The study of the interactions of the GLB-inhibitor in the Beta-Lactamase active site
The GLB compound called: Glycylboronate (Figure 1) is considered as one of the best inhibitors of the Beta- Lactamase with an IC50 equals to 9.3 nM [14,15].
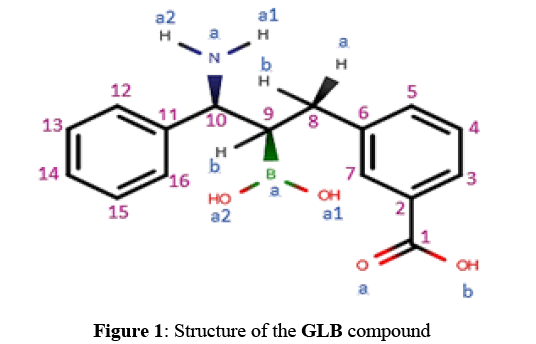
Figure 1: Structure of the GLB compound
Compared to the bibliographical references outlined above, none of these references put forward the interaction mode of this compound towards the Beta-Lactamase active site. Thus, it is interesting to consider a deeper and more advanced study in silico concerning the inhibition mechanisms set up by this compound in order to better rationalize the conception of new inhibitors towards the studied target: the Beta-Lactamase.
In fact, the reconstruction of the Beta-Lactamase-GLB complex by the molecular docking with Surflex, confirms the experimental data by giving a score equals to 4.51. The visual analysis shows a good positioning of this compound in the catalytic cavity of the enzyme (Figure 2).
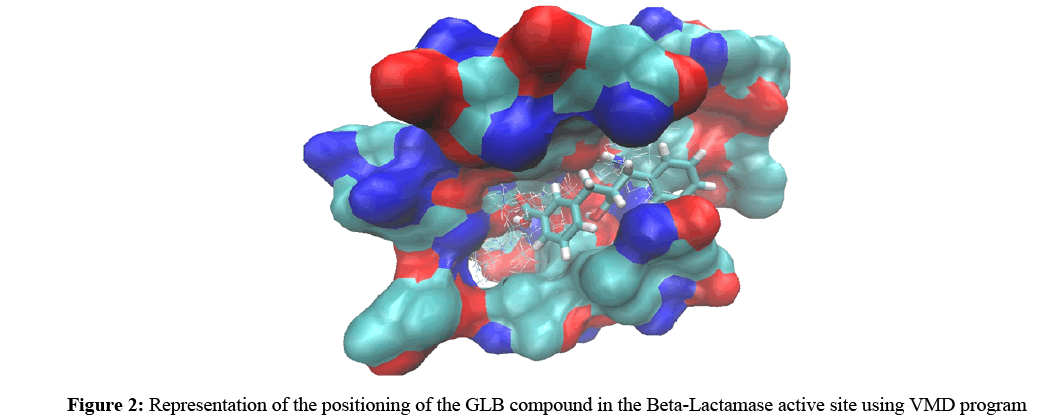
Figure 2: Representation of the positioning of the GLB compound in the Beta-Lactamase active site using VMD program
We notice as well, the formation up of 7 hydrogen bonds during the formation of Beta-Lactamase–GLB complex (Figure 3) indeed, the first oxygen of the carboxylic function carried by the C2 of the inhibitor forms two hydrogn bonds with the amine function of the residue Gly344 (distance=3.06Å) on one hand and the amine function related to the C-alpha of the residue Ser345 (distance=3.17Å) on the other hand. On its part, the second oxygen of the same carboxylic function forms another hydrogen bond with the amine function which belongs to the radical of the residue Thr343 (distance=2.97 Ǻ). The hydroxyl of the radical of the residue Ser90 undertakes two hydrogen bonds with the hydroxyls carried on the carbons in the positions C9a1 and C9a2 (distances=2.99 Ǻ and 2.91 Ǻ, respectively). The sixth hydrogen bond is found between the hydroxyl in the position C9a2 and the amine function of the radical of the residue Asn179 (distance=2.90Å). Finally, the amine function in the position C10a of the inhibitor forms a last hydrogen bond with the hydroxyl of the radical of the residue Ser345 (distance=1.93Å).
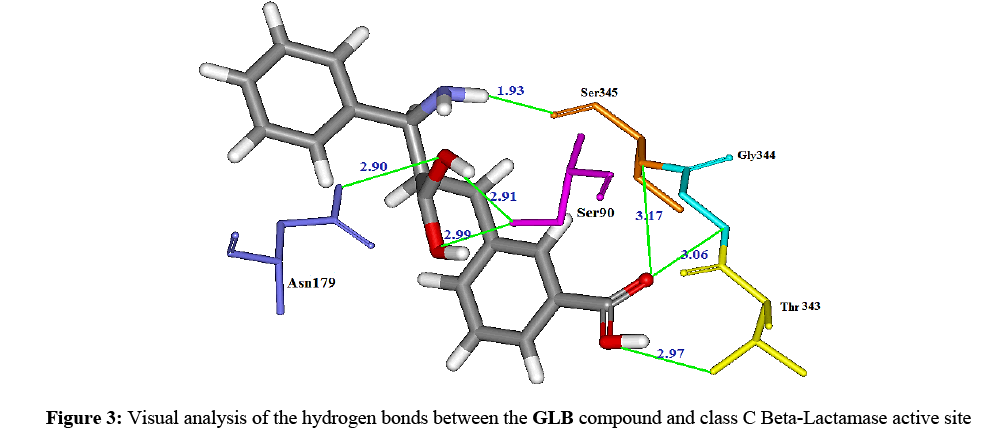
Figure 3: Visual analysis of the hydrogen bonds between the GLB compound and class C Beta-Lactamase active site
Highlighting a new beta-lactamase inhibitor
In order to suggest new potent Beta-Lactamase inhibitors, we have taken the GLB compound as a starting structure on which we have made structural modifications by introducing new functional chemical groupings. We can precisely state that the added groupings represent the ability to form hydrogen bonds with the active site of the enzyme.
Interactions between GLB-derivatives compounds and Beta-Lactamase binding site were studied using Surflex docking’s program. The results are shown in Table 1. It could be observed that the best score was obtained in the case of the compound GLB-tri-substituted. Indeed, the introduction in the position C3, of a benzene- 3’-COOH, of an -OH in the position C8b and an -COOH in the position C12 improves clearly the score, that goes from 4.51 in the case of the starting compound (GLB) to 7.91 for the new compound called GLB-tri-substituted (Figure 4).
| GLB compound | 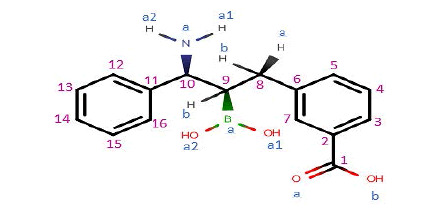 |
|||||
| GLB-derivative grouping positions | C3 | C7 | C8a | C8b | C12 | Surflex Score (Affinity) |
| GLB-compound | 4.51 | |||||
| Compound 1 | COOH | 5.98 | ||||
| Compound 2 | CHO | 6.08 | ||||
| Compound 3 | COOH | COOH | 6.37 | |||
| Compound 4 | OH | COOH | 6.67 | |||
GLB-tri-substituted  |
OH | COOH | 7.91 | |||
Table 1: Structures and docking’s results of several GLB-derivatives in the binding site of Beta-lactamase
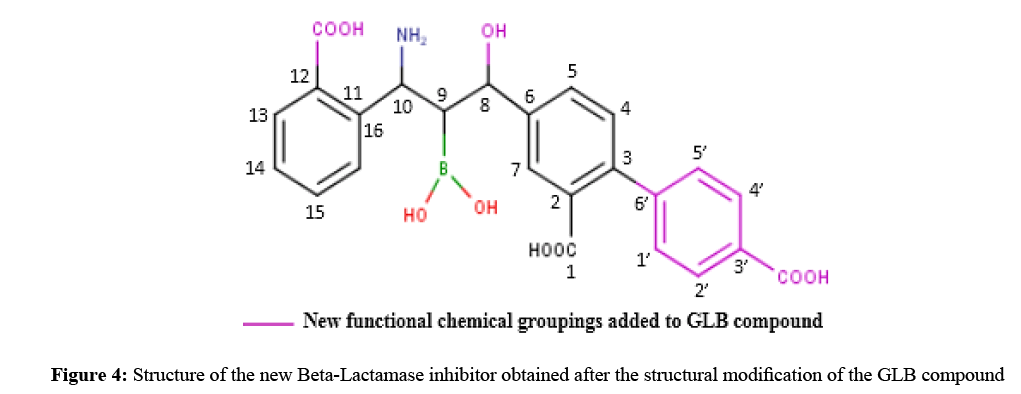
Figure 4: Structure of the new Beta-Lactamase inhibitor obtained after the structural modification of the GLB compound
Considering as the most promising inhibitor, we selected the GLB-tri-substituted compound for interaction study. Indeed, the visual analysis shows that this compound is correctly positioned and establishing eight (08) hydrogen bonds with the binding site of Beta-lactamase (Figures 5 and 6).
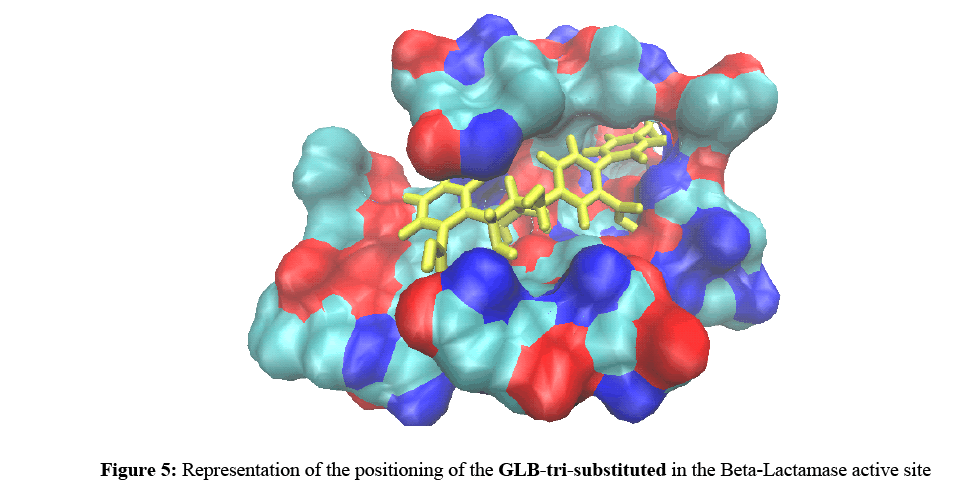
Figure 5: Representation of the positioning of the GLB-tri-substituted in the Beta-Lactamase active site
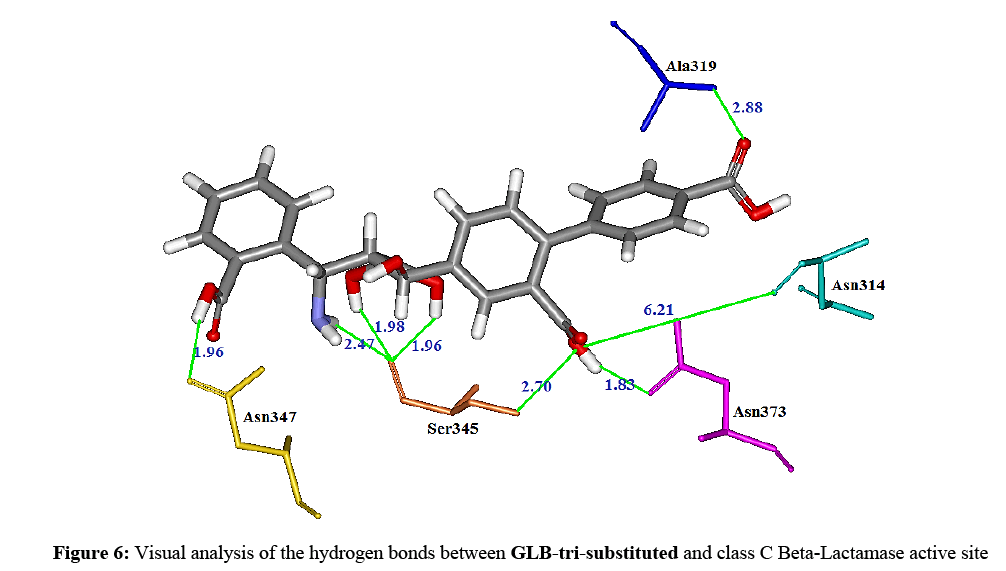
Figure 6: Visual analysis of the hydrogen bonds between GLB-tri-substituted and class C Beta-Lactamase active site
The oxygen in the position C1b the inhibitor forms three hydrogen bonds: the first one is noticed with the hydroxyl of the radical of Ser345 (distance=2.70 Å), the second one is observed with the oxygen related to the C-alpha of the residue Asn314 (distance=6.21 Å), and the third one is formed with the oxygen of the residue Asn373 (distance=1.83 Å).
The first oxygen carried by the boron in the position C9a1 forms a hydrogen bond with the oxygen related to the C-alpha of the residue Ser345 (distance=1.96 Å).
On its part, the oxygen in the position C9a2 of the inhibitor undertakes by means of another hydrogen bridge with the oxygen related to C-alpha of the residue Ser345 (distance=1.96 Å).
The amine function carried on the carbon C10 forms a hydrogen bond with the oxygen related to C-alpha of the residue Ser345 (distance=2.47 Å). The carboxylic function carried on the carbon C12 of the inhibitor forms a hydrogen bond with the oxygen that belongs to the radical of the residue Asn347 (distance=1.96 Å).
Finally, the new carboxylic funtion introduced in the position C3’ of the benzen ring in C3 of the inhibitor forms a new hydrogen bond with the nitrogen related to the C-alpha of the residue Ala319 (distance=2.88 Å).
Lipinski rule
To accomplish our study, it was important to apply the ADME filter method based on the Lipinski rule [13], in order to find out the pharmacokinetic properties (ADME) of the GLB tri-substituted stemming from the structural substitution of the Glycylboronate « GLB compound ».
The criteria of this rule have been calculated using the Molinspiration server [16]. The results are the following:
• PM=463.251 Da;
• Log P=1.22;
• Nbr AH=10;
• Nbr DH=8;
• Nb rotable bonds=9
It could be observed that the molecular weight of GLB-tri-substituted compound is less than 500 Da. The values of the hydrogen bond donors, hydrogen bond acceptors, flexible bonds and Log P value were also less than the maximum extent permitted by Lipinski’s rule. Therefore, the GLB-tri-substituted, which fulfill the five criteria of Lipinski’s rule, could be suggested as a new class C Beta-Lactamase inhibitor without causing orally administration problems [13].
The objective of this work is to contribute in the development in silico of a Beta-Lactamase inhibitor: therapeutically target confirmed, to cure the induced infections by several resisting bacteria to Beta-Lactams antibiotics especially Pseudomonas aeruginosa and Klebsiella pneumoniae.
According to that vision, we have realized structural modifications of a powerful Beta-Lactamase inhibitor known as Glycylboronate (GLB compound).
In fact, the introduction of a hydroxyl grouping in the position C8b, a carboxyl grouping of the position C12 and a benzene ring-3’-COOH has clearly improved the score that rises from 4.51 in the case of the starting compound (GLB compound) to 7.91 concerning the Glycylboronate GLB-tri-substituted. Finally, applying the Lipinski rule led us to better know that the ADME properties of the GLB-tri-substituted compound which is represented as a new inhibitor is theoretically more powerful towards the AmpC class C Beta-Lactamase that could be orally administered without causing oral administration problems.