Research Article - Der Pharmacia Lettre ( 2025) Volume 17, Issue 1
Received: 15-Jun-2024, Manuscript No. DPL-24-139081; Editor assigned: 18-Jun-2024, Pre QC No. DPL-24-139081 (PQ); Reviewed: 02-Jul-2024, QC No. DPL-24-139081; Revised: 26-Dec-2024, Manuscript No. DPL-24-139081 (R); Published: 02-Jan-2025 , DOI: 10.37532/dpl.2025.17.21
The present research work focuses on formulation, optimization and in-vivo evaluation of the orodispersible tablets of Almotriptan to achieve the desired bioavailability and give a rapid relief from the migraine headache.
Methods: The optimization was done using 22 factorial design and the formulation were subjected to several in-vitro evaluation studies. Along with stability studies for 6 months. The optimised stable formulation of Almotriptan was subjected to in-vivo studies employing animal models (rabbits) to determine the plasma concentration of the drug within required time after oral administration and the pharmacokinetic parameters such as Cmax, Tmax, AUC and others were calculated. In-vitro-in-vivo correlation was done by deconvolution using Wagner-Nelson method and the graphs were plotted to report the correlation of drugs.
Results: The almotriptan formulation F32 containing mannitol as diluent and Cross povidone 8 mg as super disintegrant has shown better release of drug i.e., 98.87% in 30 min, all the pre and post compression parameters were in limits. The optimized formulation has shown 98.27% drug release in 15 min and the same formulation was taken for in-vivo study which gave greater Cmax, Tmax and AUC values compared to marketed formulation. The in-vitro on-vivo correlation was well established with R2 value of 0.9908.
Conclusion: Almotriptan orodispersible tablets have been successfully formulated and evaluated and shows rapid release with enhanced bioavailability.
Almotriptan; Mannitol; Cross carmellose sodium; Cross povidone; Sodium starch glycolate; In-vivo study, In-vitro in- vivo correlation
Oro-dispersible tablets also called as fast dissolving or mouth dissolving tablets are those that show rapid activity by dispersing in mouth itself without any water requirement [1]. They differ from the compounds that are absorbed from other parts of gastrointestinal tract [2]. The solubilization of these drugs occurs in saliva itself which on further passage into gastro-intestine facilitates dissolution of drug [3]. Migraine is neurological disease which is characterized by mild to severe headaches that lasts from 1 hour to several hours with symptoms effecting autonomic nervous system [4]. It is a greek word meaning pain on one side of the head. Pain is caused in pulsating nature on one side of the head lasting from 2 to 72 hours [5]. Several other symptoms are associated with it like nausea, vomiting and sensitivity to sound, light [6]. Generally, migraine headache is followed by aura which is a visual, sensory or motor disturbance [7]. Triptans are the drugs that are being widely used in the present days to treat migraine other than analgesics and alkaloids [8]. The above triptan drugs selected were less explored and very few investigations have been done on them based on the literature survey [9]. Migraine headaches are to be treated immediately to get relief, so development of oral disintegrating tablets of triptans will be the apt formulation for the treatment of migraine which shows maximum effect within short time [10]. These formulations without need of water disintegrate rapidly in the mouth and shows faster absorption of drug through saliva within minutes to treat migraine headaches [11]. Hence orodispersible tablets of Antimigraine drugs were formulated using various diluents and superdisintegrants [12].
Construction of calibration curve in pH 6.8 phosphate buffer
From stock solution B 2,4,6,8 ml of solutions were pipette out and transferred into 10 ml volumetric flasks and the volume was made upto 10 ml with phosphate buffer pH 6.8 to get the solutions of concentrations 2, 4, 6 and 8 µg/ml [13]. Their absorbance was measured by U.V. spectrophotometer at their maximum wavelength analytically and calibration curve was plotted [14].
Construction of calibration curves for drugs in biological fluids.
1 ml of blank plasma and 200 µl of Almotriptan at concentrations 30 ng/ml, 60 ng/ml, 90 ng/ml, 150 ng/ml, 250 ng/ml, 300 ng/ml, 400 ng/ml and 500 ng/ml working standard solutions were transferred separately into a series of centrifugal tubes. To this added 300 µl of internal standard (Naratriptan 1000 ng/ml) and 1 ml of Acetonitrile and placed on cyclomixer for 15 sec. Then vertexes for 2 min and finally centrifuged for 5 min at 3200 rpm speed. After the centrifugation, collected the organic layer in these analytes concentrations to produce 6 ng/ml, 12 ng/ml, 18 ng/ml, 30 ng/ml, 50 ng/ml, 60 ng/ml, 80 ng/ml and100 ng/ml whereas 150 ng/ml of ISD concentration was directly injected 20 µL into HPLC
Chromatographic conditions
Mobile phase : OPA and Acetonitrile (50:50).
Flow rate : 1.0 ml/min
Column : KROMASIL, C18, 250 x 4.6 mm, 5 m.
Detector wave length : 227 nm
Injection volume : 20 mL
Run time : 10 min
Drug-excipient compatibility studies by I.R
Drug excipient compatibility studies were conducted to monitor or determine the interactions among the active pharmaceutical ingredients and all the excipients that were employed in various formulations (Table 1).
| Formulation code | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | F11 | F12 | F13 | F14 | F15 | F16 | F17 |
| Almotriptan | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 |
| SSG | 4 | 6 | 8 | 10 | 12 | 14 | 16 | ||||||||||
| CCS | 1 | 2 | 4 | 6 | 8 | 10 | |||||||||||
| CP | 4 | 6 | 8 | 10 | |||||||||||||
| Mannitol | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 | 160 |
| Aspartame | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Aerosil | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Mg stearate | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Talc | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Total weight (mg) | 176.5 | 178.5 | 180.5 | 182.5 | 184.5 | 186.5 | 188.5 | 173.5 | 174.5 | 176.5 | 178.5 | 180.5 | 182.5 | 176.5 | 178.5 | 180.5 | 182.5 |
Table 1: Composition of Almotriptan Orodispersible tablets formulated by using various concentrations of super disintegrants.
Wetting time
Wetting time is the time taken for the tablet to completely absorb water to disintegrate rapidly. A petri plate was taken and filled with certain volume of buffer solution. Filter paper or tissue paper was placed in the petri plate in the buffer solution and the tablet was placed on it. The time taken for the buffer solution to completely wet the tablet was noted. More the wetting time results more disintegration time taken by the tablet and vice versa.
Water absorption ratio
The tablets were initially weighed and the weight was noted as W1. Then they were evaluated for wetting time and the tablet weight after complete wetting was noted as W2. The water absorption ratio calculated from the formula.
R=W2-W1/W1
In-vitro disintegration studies
Disintegration time is the time in which complete tablet breaks down into finer particles and goes into the fluid. This is specific test for ODT’s as they ought to show very less disintegration time. Electrolab disintegration test apparatus was employed containing 6 tubes 3 inches long, held by 10 mesh screen and open at top. 1 tablet was placed in each tube and the basket was moved up and down in a beaker containing 1 liter pH 6.8 phosphate buffer at a height of 2.5 cm and the temperature of 37 ± 0.5°C. Then the disintegration time of tablets was noted and the average was calculated.
Drug content uniformity
The tablets formulated were tested for uniformity in drug content. The percentage of drug actually present in them compared to the incorporated drug. 20 tablets were weighed accurately and powdered, the powder equivalent to the dose of the drug was transferred to 100 ml of buffer solution and was dissolved thoroughly by shaking and was allowed to stand for 24-48 hrs. Then a sample was withdrawn, filtered through wattman filter paper and diluted suitably. The absorbance was measured from which the amount of drug was calculated. The % drug content was then calculated practically.
Dissolution studies
The dissolution/drug release studies were carried out using paddle type USP-II dissolution apparatus. pH 6.8 phosphate buffer was employed as dissolution fluid in volume of 900 ml, the rpm was maintained 50rpm, temperature was maintained at 37 ± 0.5°C. Aliquotes of 5 ml were collected at regular intervals and replaced with equal volume of fresh buffer to maintain sink conditions. The samples were filtered, suitably diluted and analysed by U.V-spectrophotometer at maximum wavelength of the drugs (Table 2).
In-vivo drug release studies
Animal dose calculation:
HED (mg/kg)=Animal dose (mg/kg) X Animal Km factor/Human Km factor.
12.5=A.D. X 37/12 =37/12*12.5/60=0.642 mg/kg
HED: Human Equivalent Dose
Animal Km factor=12, Human Km factor=37., Wt. of the rabbits=2.5 kg. So the dose of the drug taken is 1.605 mg
| S.NO | Ingredients | Quantity (mg) |
| 1 | Almotriptan | 1.605 |
| 2 | Mannitol | 30 |
| 3 | SSG | 10 |
| 4 | Aspartame | 10 |
| 5 | Aerosil | 2 |
| 6 | Mg.stearate | 4 |
| 7 | Talc | 4 |
| Total wt | 61.605 | |
Table 2: Formulation of Almotriptan orodispersible tablet for in-vivo administration.
All the ingredients were weighed accurately, passed through sieve no.22 mixed thoroughly by physical mixing and compressed into tablet by direct compression using 5 mm punch.Three marketed tablets were weighed initially and powdered, and then the powder containing the drug dose equivalent to 1.605 mg was accurately weighed and collected to administer to animals for in-vivo study.
Pharmacokinetic study: Healthy rabbits (New Zealand Albino) of either sex weighing 2.5 kg were selected and housed with CPCSEA guidelines, (Approval No.ARTI/CPCSESA/2016/ARTI19/A), (Reg.No.1722/RO/Ere/S/13/CPCSEA) were fasted overnight and had free access to drinking water.
Experimental design: Animals were separated into two experimental groups, each group consisting of three animals (n=3). The test formulation of batch (Aoptimised) was compared with (reference/ marketed formulation) with the following treatment schedule under fasted condition
Group I-Almotriptan marketed formulation.
Group II-Almotriptan optimised formulation used as test.
The test formulation of Almotriptan was formulated into orodispersible tablet by direct compression method, taking the animal dose of 1.605 mg into consideration. This is placed in the oral cavity of rabbit using tweezers and was held until the tablet has completely disintegrated. Blood samples (each of about 1-2 ml from each animal) were withdrawn from marginal ear vein at regular time intervals after administration. The collected blood samples were immediately centrifuged at 5000 rpm in ultra cooling centrifuge for 10 min at 40C. The supernatant plasma sample was separated and stored in a clean screw capped 5 ml polypropylene plasma tubes at -20°C in a deep freezer, until further analysis.
Estimation of drug from rabbit plasma
The stored plasma samples were processed at room temperature, 250 µl of plasma was added to 500 µl of Acetonitrile to precipitate the proteins. The samples were vortexed on vortex mixer for 15 min, followed by centrifugation at 10000 rpm for 15 min. The respective samples were injected into the HPLC C18 column. Injection volume was 20 µl and run time is 10 minutes.
Data analysis: The total area under plasma concentration time curve (AUC0-∝), the maximum plasma concentration (Cmax), and time to reach the maximum plasma concentration (Tmax) were selected as parameters for pharmacokinetic evaluation. The Cmax and Tmax were obtained directly from the experimental data of plasma concentration versus time. AUC0-∝ was obtained by adding the AUC (0-t) + AUC (0-∞), which was calculated by the trapezoidal rule. The differences in average of data were compared by sample analysis of variance (oneway analysis of variance) or independent sample t test. The significance of the difference was determined at 95% confident limit (P=0.05) (Figures 1-11 and Tables 3-6).
In-vitro in-vivo correlation study: (IVIVC)
Deconvolution: Level A correlation is developed by deconvolution process that converts output (plasma concentration profile) to input in-vivo dissolution of dosage form.
Wagner-Nelson method was employed in the present study for conversion. This method is used for one compartment model and is less complicated. The fraction of drug absorbed at time t (Ft) can becaluculated from the formula
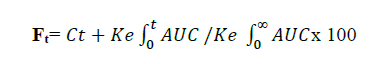
Where Ct is plasma concentration at time t,
Ke is elimination rate constant
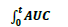 is area under curve from time zero to time t.
is area under curve from time zero to time t.
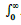 is area under curve from time zero to infinite time
is area under curve from time zero to infinite time

Figure 1: Standard calibration curve of Almotriptan pure drug in pH 6.8 Phosphate buffer.

Figure 2: Standard calibration curve of Almotriptan in spiked rabbit plasma

Figure 3: IR spectrum of pure drug and optimised formulation of Almotriptan.
|
F code |
Average Weight |
Thickness |
Hardness |
Friability (%) |
Disintegration time(sec) |
% of drug content |
Wetting time |
Water absorption ratio |
|
(mm) |
(kp) |
(sec) |
||||||
|
F1 |
176.6 ± 0.034 |
4.32 ± 0.054 |
3.76 ± 0.054 |
0.19 ± 0.075 |
38 ± 0.061 |
98.54 ± 0.034 |
19 ± 0.065 |
0.18 ± 0.054 |
|
F2 |
179.8 ± 0.051 |
4.34 ± 0.048 |
3.78 ± 0.019 |
0.19 ± 0.065 |
35 ± 0.019 |
98.32 ± 0.065 |
25 ± 0.017 |
0.193 ± 0.062 |
|
F3 |
180.5 ± 0.068 |
4.36 ± 0.014 |
3.79 ± 0.064 |
0.2 ± 0.081 |
34 ± 0.038 |
99.09 ± 0.069 |
30 ± 0.058 |
0.201 ± 0.035 |
|
F4 |
182.6 ± 0.078 |
4.4 ± 0.054 |
3.8 ± 0.058 |
0.21 ± 0.033 |
31 ± 0.041 |
97.14 ± 0.081 |
24 ± 0.049 |
0.211 ± 0.095 |
|
F5 |
184.2 ± 0.092 |
4.42 ± 0.065 |
3.81 ± 0.045 |
0.17 ± 0.021 |
25 ± 0.022 |
99.21 ± 0.027 |
22 ± 0.039 |
0.22 ± 0.0.58 |
|
F6 |
186.7 ± 0.064 |
4.43 ± 0.083 |
3.82 ± 0.097 |
0.13 ± 0.063 |
23 ± 0.077 |
99.82 ± 0.036 |
20 ± 0.058 |
0.231 ± 0.024 |
|
F7 |
188.7 ± 0.046 |
4.45 ± 0.091 |
3.83 ± 0.087 |
0.22 ± 0.035 |
22 ± 0.064 |
99.86 ± 0.022 |
18 ± 0.044 |
0.298 ± 0.048 |
|
F8 |
173.2 ± 0.055 |
4.46 ± 0.062 |
3.84 ± 0.027 |
0.19 ± 0.046 |
42 ± 0.045 |
99.79 ± 0.011 |
29 ± 0.085 |
0.17 ± 0.057 |
|
F9 |
175.4 ± 0.042 |
4.49 ± .0.033 |
3.86 ± 0.035 |
0.21 ± 0.044 |
40 ± 0.033 |
99.55 ± 0.054 |
26 ± 0.069 |
0.185 ± 0.034 |
|
F10 |
177.1 ± 0.036 |
4.28 ± .0.044 |
3.74 ± 0.064 |
0.18 ± 0.088 |
39 ± 0.086 |
99.71 ± 0.062 |
23 ± 0.077 |
0.216 ± 0.066 |
|
F27 |
179.7 ± 0.058 |
4.3 ± 0.026 |
3.75± .0074 |
0.19± 0.071 |
37 ± 0.024 |
98.12 ± 0.086 |
28 ± 0.055 |
0.21 ± 0.092 |
|
F11 |
182.4 ± 0.045 |
4.31 ± .0.065 |
3.75 ± 0.064 |
0.22 ± 0.062 |
38 ± 0.051 |
98.22 ± 0.049 |
20 ± 0.061 |
0.24 ± 0.084 |
|
F12 |
181.8 ± 0.024 |
4.34 ± .0.055 |
3.77 ± 0.038 |
0.24 ± 0.014 |
39 ± 0.059 |
99.07 ± 0.057 |
25 ± 0.073 |
0.303 ± 0.057 |
|
F13 |
177.4 ± 0.065 |
4.31 ± 0.061 |
3.75 ± 0.022 |
0.2 ± 0.074 |
37 ± 0.064 |
98.42 ± 0.036 |
22 ± 0.026 |
0..176 ± 0.021 |
|
F14 |
179.3 ± 0.087 |
4.34 ± 0.075 |
3.76 ± 0.021 |
0.21 ± 0.089 |
36 ± 0.077 |
98.14 ± 0.081 |
19 ± 0.044 |
0.181 ± 0.066 |
|
F15 |
180.9 ± 0.049 |
4.36 ± 0.051 |
3.77 ± 0.054 |
0.22 ± 0.028 |
37± 0.068 |
99.17 ± 0.055 |
24 ± 0.017 |
0.217 ± 0.054 |
|
F16 |
183.7 ± 0.035 |
4.4 ± 0.034 |
3.79 ± 00.2 |
0.24 ± 0.06 |
39 ± 0.054 |
97.89 ± 0.06 |
21 ± 0.06 |
0.22 ± 0.025 |
Table 3: Data of postcompression parameters of orodispersible tablets of Almotriptan.

Figure 4: In-vitro dissolution data observed from Almotriptan orodispersible tablets formulated with various super disintegrants.
|
F17 |
24.31 ± 0.045 |
35.24 ± 0.026 |
0.8873 |
0.9511 |
0.0285 ± 0.012 |
50.30 ± 0.162 |
|
F18 |
20.54 ± 0.017 |
34.55 ± 0.019 |
0.7812 |
0.9623 |
0.0337 ± 0.006 |
44.56 ± 0.057 |
|
F19 |
15.82 ± 0.062 |
25.27 ± 0.044 |
0.6862 |
0.9512 |
0.0438 ± 0.006 |
42.63 ± 0.115 |
|
F20 |
11.12 ± 0.022 |
24.32 ± 0.036 |
0.6687 |
0.9541 |
0.0623± 0.007 |
40.14 ± 0.066 |
|
F21 |
9.66 ± 0.026 |
19.54 ± 0.022 |
0.7423 |
0.9632 |
0.0717 ± 0.002 |
41.05 ± 0.148 |
|
F22 |
9.52 ± 0.087 |
19.12 ± 0.044 |
0.8129 |
0.9762 |
0.0727 ± 0.004 |
41.3 ± 0.241 |
|
F23 |
9.51 ± 0.066 |
19.06 ± 0.052 |
0.7582 |
0.9592 |
0.0728 ± 0.003 |
41.38 ± 0.062 |
|
F24 |
29.67± 0.034 |
40.58 ± 0.024 |
0.6944 |
0.9629 |
0.0233 ± 0.001 |
37.36 ± 0.081 |
|
F25 |
29.54 ± 0.011 |
39.87 ± 0.078 |
0.6337 |
0.9539 |
0.0234± 0.002 |
36.4 ± 0.254 |
|
F26 |
26.88 ± 0.047 |
39.52 ± 0.022 |
0.7891 |
0.9534 |
0.0257± 0.005 |
37.5 ± 0.068 |
|
F27 |
25.11 ± 0.061 |
34.66 ± 0.031 |
0.8048 |
0.9661 |
0.02759 ± 0.007 |
33.94 ± 0.158 |
|
F28 |
19.43 ± 0.033 |
29.43 ± 0.055 |
0.6732 |
0.9683 |
0.0356± 0.003 |
33.65 ± 0.324 |
|
F29 |
14.79 ± 0.084 |
29.42 ± 0.042 |
0.7422 |
0.9565 |
0.0468± 0.002 |
34.09 ± 0.245 |
|
F30 |
14.88 ± 0.095 |
30.05 ± 0.019 |
0.6892 |
0.9757 |
0.0465 ± 0.004 |
41.12 ± 0.085 |
|
F31 |
14.67 ± 0.067 |
25.88 ± 0.048 |
0.8115 |
0.9683 |
0.0472 ± 0.006 |
34.03 ± 0.073 |
|
F32 |
14.56 ± 0.025 |
25.1 ± 0.028 |
0.7446 |
0.9884 |
0.0476 ± 0.005 |
34.2 ± 0.142 |
|
F33 |
13.78 ± 0.034 |
25.07 ± 0.027 |
0.8552 |
0.9782 |
0.0503 ± 0.003 |
34.3 ± 0.321 |
Table 4: In-vitro release kinetics data observed fromAlmotriptan orodispersible formulations

Figure 5: Chromatogram of blank plasma for Almotriptan in-vivo studies by HPLC method.

Figure 6: Chromatogram of internal standard sumatriptan for Almotriptan in-vivo studies in rabbit plasma by HPLC method.

Figure 7: Chromatogram of linearity data of Almotriptan pure drug in rabbit plasma by HPLC for maximum concentration.

Figure 8: Chromatogram of optimised formulation of Almotriptan in rabbit plasma by HPLC.

Figure 9: Chromatogram of marketed formulation of Almotriptan in rabbit plasma by HPLC.
| Time (hrs) | Optimized (ng/ml) | Marketed (ng/ml) |
| 0 | 0 | 0 |
| 1 | 25.46 ± 1.28 | 19.18 ± 1.12 |
| 1.5 | 33.42 ± 5.28 | 25.26 ± 1.22 |
| 2 | 44.17 ± 3.04 | 35.13 ± 1.16 |
| 2.5 | 48.72 ± 2.11 | 40.18 ± 1.08 |
| 3 | 43.84 ± 1.24 | 30.29 ± 1.04 |
| 6 | 29.42 ± 1.10 | 20.56 ± 1.16 |
| 8 | 22.39 ± 1.56 | 15.14 ± 1.26 |
| 10 | 18.52 ± 1.08 | 12.52 ± 1.20 |
| 16 | 11.32 ± 1.84 | 8.54 ± 1.16 |
| 24 | 7.54 ± 2.26 | 5.32 ± 1.22 |
Table 5: In-vivo plasma concentration Vs. time data of Almotriptan optimised formulation.

Figure 10: Comparative plasma concentration vs. time curves of Almotriptan optimized and marketed orodispersible formulation.
| Parameters | Optimized | Marketed | t-test ‘p’ value |
| Cmax(ng/ml) | 48.72 ± 0.04 | 40.18 ± 0.24 | 0.0024 |
| T(max) hrs | 2.5 ± 0.26 | 2.5 ± 0.02 | 0.0035 |
| Vd (litres) | 185.5 ± 0.015 | 162.93 ± 0.056 | 0.0047 |
| Clearance (Lhr-1) | 56.44 ± 0.234 | 48.32 ± 0.137 | 0.0044 |
| AUC (0) ng.h/ml | 492.15 ± 0.12 | 336.22 ± 0.16 | 0.0018 |
| MRT (hrs) | 6.363 ± 0.278 | 7.108 ± 0.158 | 0.0029 |
| KE (hr-1) | 1.270 ± 0.033 | 0.926 ± 0.021 | 0.0016 |
| T50 (hr) | 0.545 ± 0.026 | 0.748 ± 0.054 | 0.0036 |
| T90 (hr) | 1.804 ± 0.047 | 2.07 ± 0.044 | 0.0027 |
Table 6: Pharmacokinetic parameters determined by in-vivo studies on Almotriptan optimized and marketed formulation.

Figure 11: In-vitro in-vivo correlation values of in-vitro drug release and fraction of drug absorbed in-vivo of Almotriptan formulation.
Then the calibration curve was constructed in ph 6.8 phosphate buffer and spiked rabbit plasma by using the various dilute concentrations of Almotriptan and the R2 value was found to be 0.999 which shows that linearity exists between the values. The drug excipient compatibility done by IR spectroscopy shows no alteration in the functional peaks for both pure drug and optimised formulation and there is no interaction between the drug and excipients used in the formulations. The post compression parameters were also found to be within IP limits and satisfactory. The drug content uniformity was found to be above 97% for all the formulations and hence there is no wastage of drug dose and complete dose will be available to the body. The lesser wetting time and higher water absorption ratio also results in rapid uptake of fluid and rapid disintegration of tablets. Among all the formulations containing various superdisintegrants F7 was found to show the best relase of 100% in 20 min and is the optimized formulation which has mannitol as diluent and SSG (8%) as superdisintegrant. Drug kinetic parameters were determined by all the formulations such as T50, T90, R2, KE and dissolution effeciency and statistical evaluation of ANOVA and t-test was performed on those parameters where the ‘p’ value was less than 0.05. It shows that there is no significant variation between the rate constant and dissolution effeciency of the formulation and the drug release studies conducted were efficient. All the pharmacokinetic parameters determined were better in case of the optimised formulation compared to marketed formulation and there was in-vitro in-vivo correlation found and hence the bioavailability of Almotriptan can be enhanced if it is formulated as orodispersible tablet.
The orodispersible tablets of Almotriptan were successfully formulated by employing various superdisintegrants in varying concentrations and evaluated for precompression and post compression parameters, optimized and the stable fomulation has shown good in-vivo profile and correlation.
I would like to thank my guide Dr. T.E.Gopala Krishna murthy for his invaluable guidance constant support and faith in me throughout the research. I would like to thank my co-guide Dr. A.Srinivasa Rao for his share of knowledge and valuable inputs in to the research. I would like to thank my fellow faculty for their support and availability.
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]
[Crossref] [Google Scholar] [PubMed]